


The mutations that drive cancer formation are often found in “hub” genes that regulate many aspects of cell growth and survival. But these key genes are not always good therapeutic targets — some are even considered “undruggable.” In the latest issue of GENETICS, Bailey et al. identify a strategy for fighting cancer cells that carry a mutation in one such difficult-to-target hub gene, FBW7. This tumor suppressor is mutated in approximately 6% of all cancer cases, including around a third of cases of T-cell acute lymphocytic leukemia and cholangiocarcinoma.
The FBW7 protein is part of the SCF ubiquitin ligase complex that marks other proteins for selective degradation. Many of these substrates are involved in oncogenesis (cancer formation), including the cell cycle regulator cyclin-E and the cell death inhibitor MCL1. Without FBW7 keeping them in check, these cancer-promoting proteins accumulate in the cell and can trigger uncontrolled division.
Even though FBW7’s tumor suppressor function is well understood, little is known about how the mutant cells might be targeted for treatment. That’s because inactivating the function of a tumor suppressor gene (e.g. with a drug) would encourage, not inhibit cancer, while many of FBW7’s oncogenic substrates are themselves difficult to manipulate with pharmaceuticals.
The authors approached this problem by screening for “synthetic lethal” partners of FBW7, which are proteins needed for cell survival when FBW7 is non-functional. They screened approximately 16,000 human genes by knocking down their expression in fbw7 mutant colorectal cancer cells and FBW7 wild-type cells, looking for proliferation effects that were specific to the mutants.
One of the candidates identified in the screen was BUBR1, which encodes a component of the mitotic spindle assembly checkpoint (SAC). This checkpoint prevents cell division from proceeding until all chromosomes are correctly attached to the spindle apparatus. When BUBR1 expression is knocked down in fbw7 mutant cells, the cells proliferate slower than wild-type and become more prone to losing and gaining chromosomes through cell division errors. This vulnerability of fbw7 cells was confirmed by tests with other SAC components.
Why do fbw7 cells have such a critical need for spindle assembly surveillance? One part of the answer is that the cells have dysregulated cyclin E. This is suggested by the fact that dampening expression of cyclin E rescues fbw7 mutant cells from their dependence on BUBR1. Cyclin E is not the whole story, however, because boosting levels of this protein alone was not enough to make FBW7 wild-type cells sensitive to SAC loss; this was only achieved by overexpressing another FBW7 substrate, MCL1, along with a form of cyclin E sometimes seen in cancer cells.
The results show that in this cell culture model, fbw7 mutant cells depend on the SAC for their cancerous potential, likely because they have cell cycle defects that necessitate extra time for spindle assembly. Without this breathing room, the dividing cells may be more prone to lethal chromosome segregation mistakes.
The authors suggest that other cancer cell types — many of which experience chronic chromosome instability — might also depend on the SAC. They suggest that exploiting this weak spot could be a promising strategy for developing anticancer drugs, especially since blocking SAC function could have fewer side-effects than existing anti-mitotic drugs. Though there is a long way to go before this idea could be applied in the clinic, the “undruggable” target FBW7 may have pointed the way to a more accessible chink in cancer’s armor.
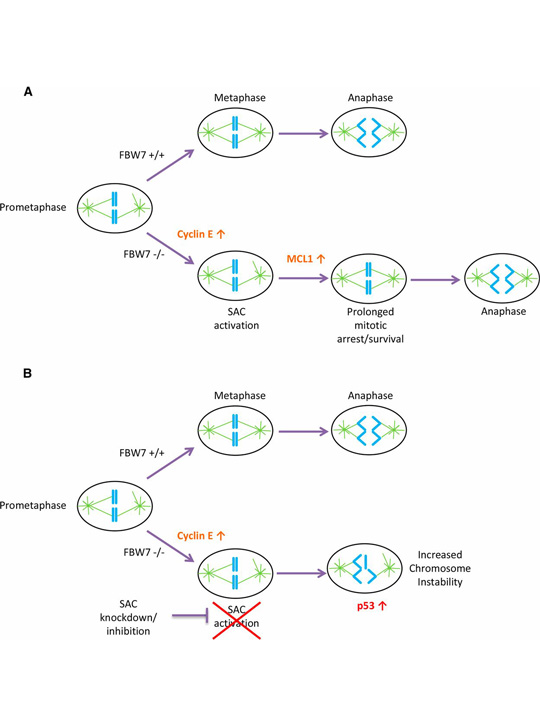
A model for SAC dependence in cells lacking FBW7. From Baikey et al. (A) FBW7 +/+ cells in prometaphase efficiently align their chromosomes and correctly segregate properly in anaphase with less reliance on the SAC. FBW7 −/− cells have an increase in cyclin E, which causes problems in mitosis and may lead to SAC activation. FBW7 −/− cells can survive prolonged SAC activation in part because of stabilization of MCL1 allowing more time for chromosome alignment. (B) A decrease in SAC activation by knockdown of BUBR1 may significantly shorten the time available for chromosome alignment in FBW7 −/− cells, resulting in improper chromosome segregation and intolerable levels of chromosome instability.
Citation:
Bailey, M. L., Singh, T., Mero, P., Moffat, J., & Hieter, P. (2015). Dependence of Human Colorectal Cells Lacking the FBW7 Tumor Suppressor on the Spindle Assembly Checkpoint. Genetics, 201(3), 885-895. Doi: 10.1534/genetics.115.180653
Cristy Gelling is a science writer, lapsed yeast geneticist, and former Communications Director at the GSA.
View all posts by Cristy Gelling »Read more in
-

Early Career Leadership Spotlight: Julio Molina Pineda
We’re taking time to get to know the members of the GSA’s Early Career Scientist Committees. Join us to learn more about our early career scientist advocates. Julio Molina Pineda Policy and Advocacy University of Arkansas Research Interest My research interests focus on using model organisms to genetically dissect complex traits related to human disease. My…
-

Early Career Leadership Spotlight: Peiwei Chen
We’re taking time to get to know the members of the GSA’s Early Career Scientist Committees. Join us to learn more about our early career scientist advocates. Peiwei Chen Accessibility Subcommittee California Institute of Technology Research Interest Far from a harmonious place, the genome is a battleground, where every bit of DNA fights for inheritance and…
-

#Dros23 GSA Poster Award winners
We are pleased to announce the GSA Poster Award winners from the 64th Annual Drosophila Research Conference! Undergraduate and graduate student members of the GSA were eligible for the awards, and a hard-working team of postdocs volunteered their time as judges. Congratulations to all! Undergraduate Students 1st Place: Sofia Karter Lopez, University of Toronto “Rab11 mediates E-cadherin recycling during…
-

Congratulations to the Fall 2022 DeLill Nasser Awardees!
GSA is pleased to announce the recipients of the DeLill Nasser Award for Professional Development in Genetics for Fall 2022! Given twice a year to graduate students and postdoctoral researchers, DeLill Nasser Awards support attendance at meetings and laboratory courses. The award is named in honor of DeLill Nasser, a long-time GSA supporter and National Science Foundation…
-

New editors join GENETICS, G3 editorial boards
Several new editors are joining the GSA Journals. We’re excited to welcome Ricardo Zayas to the GENETICS editorial board under the Molecular Genetics of Development section, and on the G3: Genes|Genomes|Genetics board, we welcome Polly Campbell, Kevin Vogel, Joe Parker, and Ricardo Mallarino. Ricardo Zayas Associate Editor Ricardo Zayas is a Professor of Biology at…
-

Worms and Flies Provide Key Clues to Medical Mystery
This article is part of a series of posts outlining the history and impact of research in experimental organisms. The series is developed in collaboration with the GSA Public Communications and Engagement Committee. By the time Bertrand Might was six months old, it was clear something was amiss. His muscles weren’t developing normally; he was…
-

Congratulations to the 2023 Early Career Leadership Program Cohort!
The Genetics Society of America (GSA) is excited to announce the latest cohort of student, postdoc, and early-career research leaders joining the Early Career Leadership Program. Participants receive training and mentoring while serving on committees charged with understanding the needs, interests, concerns, and challenges of early career scientist members of the GSA. As part of…
-

GSA LOCI: Local Outreach Community Initiatives @ GSA Conferences
Highlights: Local Outreach Community Initiatives (LOCI): The Genetics Society of America is committed to supporting the communities of the host cities of our conferences. This new year, we are excited to reconnect with our GSA community in meaningful ways within and beyond our existing programming. The GSA membership has created a caring and supportive environment…
-

New members of the GSA Board of Directors: 2023–2025
We are pleased to announce the election of five new leaders to the GSA Board of Directors: 2023 Vice President/2024 President Mariana Wolfner Distinguished Professor of Molecular Biology and Genetics and Stephen H. Weiss Presidential Fellow My research has focused on the genes and pathways that mediate sexual development and reproduction, primarily in Drosophila. From…
-

Lance David Miller: Lighting Your Own Fire by Finding the Right Resources
By Daniel J. Gironda In the Paths to Science Policy series, we talk to individuals who have a passion for science policy and are active in advocacy through their various roles and careers. The series aims to inform and guide early career scientists interested in science policy. This series is brought to you by the…
-

Graça Almeida-Porada: The Importance of Communication in a Technologically Advancing World
By Daniel J. Gironda In the Paths to Science Policy series, we talk to individuals who have a passion for science policy and are active in advocacy through their various roles and careers. The series aims to inform and guide early career scientists interested in science policy. This series is brought to you by the…