

Guest post by Kathryn “Kaylee” Mueller, Phase Genomics.
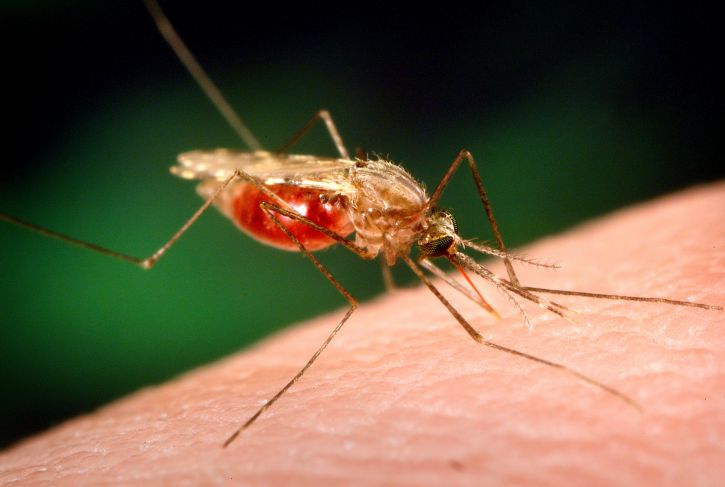
Plasmodium parasites—the microbes that cause malaria—are right at home in the tropics. After all, tropical regions harbor the two animals that the malaria parasites need to complete their complex lifecycle: female Anopheles mosquitoes and human beings. And in 2017 alone, Plasmodiumracked up 219 million cases of malaria, with 435,000 deaths.
Though malaria has spread over time across the world’s tropical areas, one region in particular—sub-Saharan Africa—unquestionably remains ground zero for the disease. African countries accounted for more than 90 percent of malaria cases and malaria deaths in 2017, according to the World Health Organization. Plasmodium‘s options for mosquito hosts in sub-Saharan Africa may explain malaria’s outsized presence there.
“Africa suffers disproportionately,” says Nora Besansky, a malaria researcher at the University of Notre Dame. “This is mainly due to the dominance of two highly efficient mosquito vectors of human malaria found throughout that region: Anopheles gambiae and Anopheles funestus.”
The twin scourges of malaria in Africa
Researchers long paid attention to An. gambiae due to its knack for transmitting the deadliest of the Plasmodium species. But scientists were slower to recognize that An. funestus, the mosquito that shares much of An. gambiae‘s range, also shows grim aptitude as a malaria vector. For example, An. funestus strongly prefers to bite humans over other animals. In addition, it tends to hang out indoors, gaining more opportunities to bite people and potentially spread malaria.
A game of catch-up
Yet scientists know much less about An. funestus than An. gambiae. Laboratory strains of An. funestus have been around for just 19 years, compared to more than 65 years for An. gambiae. The An. gambiae genome was released in 2002, the second for insects. The An. funestus genome came out in 2015, but its fragmented assembly consists of almost 1,400 scaffolds—despite a nuclear genome of just three chromosome pairs—resulting in fragmented genes, missing gene clusters, and little reliable structural information about the genome.
Assembling a better genome
Besansky is part of a team that decided to start over. They have produced a new, chromosome-scale assembly of the An. funestus genome—all from a pool of individuals of an An. funestus laboratory strain. The team used PacBio sequencing to produce long-read sequences, which they assembled into larger contigs and then filtered to remove duplicated sequences.
“Since this was a colony of mosquitos, there was high sequence duplication present in the data,” said Jay Ghurye, a member of the team and doctoral student at the University of Maryland, College Park. “We had to remove the duplications before scaffolding them using Hi-C data.”
That Hi-C data came from Phase Genomics, a partner in this project. Phase Genomics used its Proximo Hi-C approach to collapse the team’s contigs into longer scaffolds and—ultimately—produce three chromosome-length assemblies for An. funestus.
A complete roadmap
Based on contig and scaffold length alone, the new genome for An. funestusis a 100-fold improvement on the prior assembly, according to the researchers.
“Just as driving from New York to Los Angeles is facilitated by a roadmap, genetic research also is more powerful, efficient, and accurate if the 260 million puzzle pieces of nucleotides in the nuclear genome are properly ordered and oriented,” said Dr. Besansky.
The team hopes that this more complete picture of the An. funestus genome will help scientists understand what makes it such an effective malaria vector and come up with measures to countermand it.
One large to-do list for one little bug
Researchers already have a bevy of burning questions about An. funestus.
For instance, according to the World Health Organization, An. funestus is fast evolving resistance to the pyrethroid insecticides that are used to treat bed nets. The new genome includes features left out of the first assembly, such as genes arranged in tandem clusters. And at least one type of gene often found in these types of clusters—cytochrome P450 genes—has already been implicated in pyrethroid resistance in An. funestus.
Resistance is spreading in other ways too. Some An. funestus populations may have evolved behavioral adaptations to evade anti-mosquito controls. Scientists can now employ the new genome to help locate the genetic underpinnings of these traits.
With the new genome, researchers should also have an easier time detecting chromosomal rearrangements, such as inversions, among An. funestus populations.
“Inversions affect the spread of malaria, indirectly if not directly, because they typically contain hundreds or thousands of genes involved in climatic or local adaptation—allowing their mosquito carriers to fully exploit heterogeneous and otherwise challenging environments,” says Besansky.
An. funestus has obviously been busy, employing multiple approaches to dodge anti-mosquito measures. But all of these adaptations originate from the same source: the An. funestus genome. With that toolbox now completely sequenced, aligned, assembled and annotated, scientists may, at last, have the right blueprints to bring this malaria vector to heel.
Guest posts are contributed by members of our community. The views expressed in guest posts are those of the author(s) and are not necessarily endorsed by the Genetics Society of America. If you'd like to write a guest post, e-mail jtreboschi@genetics-gsa.org.
View all posts by Guest Author »Read more in
-

Early Career Leadership Spotlight: Julio Molina Pineda
We’re taking time to get to know the members of the GSA’s Early Career Scientist Committees. Join us to learn more about our early career scientist advocates. Julio Molina Pineda Policy and Advocacy University of Arkansas Research Interest My research interests focus on using model organisms to genetically dissect complex traits related to human disease. My…
-

Early Career Leadership Spotlight: Peiwei Chen
We’re taking time to get to know the members of the GSA’s Early Career Scientist Committees. Join us to learn more about our early career scientist advocates. Peiwei Chen Accessibility Subcommittee California Institute of Technology Research Interest Far from a harmonious place, the genome is a battleground, where every bit of DNA fights for inheritance and…
-

#Dros23 GSA Poster Award winners
We are pleased to announce the GSA Poster Award winners from the 64th Annual Drosophila Research Conference! Undergraduate and graduate student members of the GSA were eligible for the awards, and a hard-working team of postdocs volunteered their time as judges. Congratulations to all! Undergraduate Students 1st Place: Sofia Karter Lopez, University of Toronto “Rab11 mediates E-cadherin recycling during…
-

Congratulations to the Fall 2022 DeLill Nasser Awardees!
GSA is pleased to announce the recipients of the DeLill Nasser Award for Professional Development in Genetics for Fall 2022! Given twice a year to graduate students and postdoctoral researchers, DeLill Nasser Awards support attendance at meetings and laboratory courses. The award is named in honor of DeLill Nasser, a long-time GSA supporter and National Science Foundation…
-

New editors join GENETICS, G3 editorial boards
Several new editors are joining the GSA Journals. We’re excited to welcome Ricardo Zayas to the GENETICS editorial board under the Molecular Genetics of Development section, and on the G3: Genes|Genomes|Genetics board, we welcome Polly Campbell, Kevin Vogel, Joe Parker, and Ricardo Mallarino. Ricardo Zayas Associate Editor Ricardo Zayas is a Professor of Biology at…
-

Worms and Flies Provide Key Clues to Medical Mystery
This article is part of a series of posts outlining the history and impact of research in experimental organisms. The series is developed in collaboration with the GSA Public Communications and Engagement Committee. By the time Bertrand Might was six months old, it was clear something was amiss. His muscles weren’t developing normally; he was…
-

Congratulations to the 2023 Early Career Leadership Program Cohort!
The Genetics Society of America (GSA) is excited to announce the latest cohort of student, postdoc, and early-career research leaders joining the Early Career Leadership Program. Participants receive training and mentoring while serving on committees charged with understanding the needs, interests, concerns, and challenges of early career scientist members of the GSA. As part of…
-

GSA LOCI: Local Outreach Community Initiatives @ GSA Conferences
Highlights: Local Outreach Community Initiatives (LOCI): The Genetics Society of America is committed to supporting the communities of the host cities of our conferences. This new year, we are excited to reconnect with our GSA community in meaningful ways within and beyond our existing programming. The GSA membership has created a caring and supportive environment…
-

New members of the GSA Board of Directors: 2023–2025
We are pleased to announce the election of five new leaders to the GSA Board of Directors: 2023 Vice President/2024 President Mariana Wolfner Distinguished Professor of Molecular Biology and Genetics and Stephen H. Weiss Presidential Fellow My research has focused on the genes and pathways that mediate sexual development and reproduction, primarily in Drosophila. From…
-

Lance David Miller: Lighting Your Own Fire by Finding the Right Resources
By Daniel J. Gironda In the Paths to Science Policy series, we talk to individuals who have a passion for science policy and are active in advocacy through their various roles and careers. The series aims to inform and guide early career scientists interested in science policy. This series is brought to you by the…
-

Graça Almeida-Porada: The Importance of Communication in a Technologically Advancing World
By Daniel J. Gironda In the Paths to Science Policy series, we talk to individuals who have a passion for science policy and are active in advocacy through their various roles and careers. The series aims to inform and guide early career scientists interested in science policy. This series is brought to you by the…