

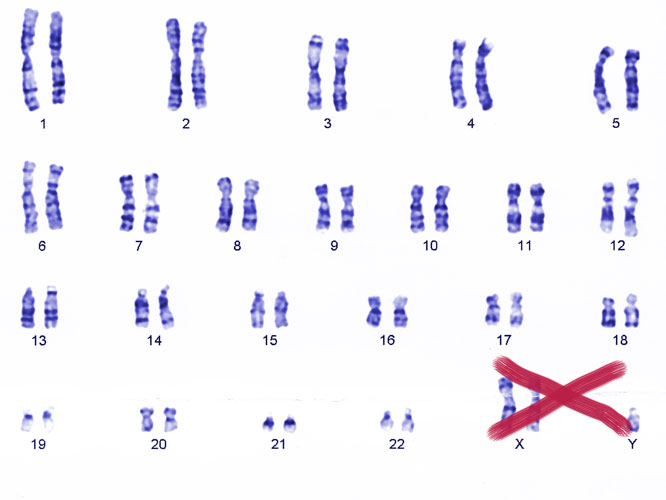
One of the major risk factors for autoimmune diseases is being born with two copies of the X chromosome. For example, women—who typically carry two Xs—face around ten times the risk of lupus, while men with lupus are around 15 times more likely than the general population to carry two Xs and a Y (Klinefelter syndrome), rather than the usual single X and Y.
The luck of the sex-chromosome draw affects many other complex diseases in prevalence, severity, symptoms, and rates of progression. Women face about twice the risk of depression, for example, while men are three to five times more prone to liver cancer. Though environmental factors like sex hormones and alcohol consumption are behind some of the differences, Alon Keinan (Cornell University) argues that genetic variation carried on sex chromosomes must also play a role.
But how big is that role? He says there is little conclusive evidence. The problem is, the sex chromosomes have been largely ignored in the flood of human genome-wide association studies (GWAS) of recent years.
“We are conducting all these genome-wide studies, but sometimes forget that the genome also includes sex chromosomes,” he says. There are many other unknowns in how sex chromosomes affect complex disease, including the population-wide variation in X-chromosome inactivation, a crucial factor in the expression of X-linked variants.
For the 2014 American Society for Human Genetics meeting in October, Keinan teamed up with Melissa Wilson Sayres (Arizona State University) to organize a session called “The X-Factor of Complex Disease: From Evolution to Association Studies of the X Chromosome” to discuss these issues and more. The idea was to bring together speakers with expertise ranging from evolutionary to functional genetics to present research on sex chromosomes and their relevance to interpretations of human medical genetic data.
“Part of the motivation is that medicine today is often driven by diagnosis and treatment of males. We would like to better understand the genetic risk factors that affect males and females differentially, to make at least one step towards sex-specific diagnosis and treatment,” says Keinan.
GSA Journals Assistant Editor Cristy Gelling caught up with Wilson Sayres and Keinan to discuss X-linked disease associations and the evolution of X-inactivation.
Bringing X back
Although the X chromosome makes up about 5% of the human genome and carries 4% of all genes, it is linked to fewer than 0.5% of associations in NHGRI’s GWAS catalog. For example, chromosome 7, similar in size to the X, has about 10 times as many associations. In model organisms, sex chromosomes are responsible for more phenotypic variation than you would predict from their size. But however you do the math, we’ve likely missed the vast majority of the X-linked associations with complex disease.
The main problem is that most GWAS discard all data from the X chromosome. “It’s understandable,” says Keinan. “You can imagine when researchers are competing to publish quickly, it’s easy to ignore something that is only 5% of the genome and needs extra time and special expertise.”
Even when the X is included, it’s typically analyzed using methods designed for autosomes. But standard statistical models for detecting significant associations don’t perform well for X chromosomes because they have unique patterns of diversity. For example, because recombination is suppressed in sex chromosomes, they evolve very differently from autosomes. One speaker at the session, Krishna R. Veeramah of Stony Brook University, compared the effects of natural selection on the X chromosomes to the autosomes in humans and apes. The X chromosomes in most of these species (including humans) show evidence for greater purifying selection and faster adaptive evolution rates compared to autosomes.
Sex-biased social practices, such as polygyny and male migration also affect variation on the X chromosome in ways that can distort association tests. One particularly recent example of a sex-biased demographic effect was caused by human slavery in the United States. Today’s African-American populations carry, on average, a higher proportion of African ancestry on their X chromosomes compared to autosomes. This shows much of the European genetic heritage of African-Americans is derived from male, rather than female ancestors—likely the legacy of white men fathering children with the women they enslaved.
It’s not only the different models of variation that create headaches when analyzing X-linked GWAS data. All the other steps of analysis, including determining an individual’s genotype, filtering out poor quality data, and imputation of missing genotype values, can be distorted by the difference in copy number between sexes. The end result of applying autosomal pipelines to the X chromosome is lower statistical power and a higher rate of false positives.
In his presentation, Keinan described a new “XWAS” analysis pipeline developed by several graduate students and postdoctoral fellows in his lab tailored to account for the biology of X chromosomes. To test the performance of the pipeline, the group used it to reanalyze existing autoimmune disease GWAS data for X-linked associations. The results replicated several previously found associations and revealed new candidate disease risk genes on the X chromosome with plausible links to immune function.
So can we now reanalyze the back-catalog of previously performed GWAS and unearth all those missing X-linked associations? Not quite. Although many GWAS datasets are available for reanalysis, they are usually deposited as processed data that have already been through quality control steps. In most cases, these QC steps have not been corrected for sex, and in some cases, the X chromosome data have even been discarded. Keinan hopes more geneticists will start depositing their GWAS results as raw data.
“All I would like to be able to do at this point is take a look at data that’s already available. A huge amount of taxpayer money has been invested in GWAS, which has paid off in many discoveries and can pay off further if the data generated is available for reanalysis,” he says.
Escape from X-inactivation
Another major complication for modelling how an X-linked variant contributes to disease is X inactivation, a process in which one of the two X-chromosomes in genetic females is randomly silenced. Which of the two chromosomes gets silenced varies from tissue to tissue and cell to cell. That means a particular X-linked variant may or may not be expressed in the tissues relevant to the disease.
X-inactivation doesn’t affect the entire chromosome; around 15 percent of genes “escape” inactivation and are expressed. Adding to this complexity, around ten percent of the genes that escape silencing only do so in some individuals and not in others. At the last talk of the session, Christine Disteche (University of Washington) presented results suggesting the genes that escape X inactivation also vary between tissues, resulting in sex-specific differences in phenotypes. But taking these phenomena into account in models of disease association is not possible because we simply don’t know enough about X-inactivation and its variation in humans, says Wilson Sayres.
“Right now, what we know about X-inactivation heterogeneity is based on nine cell lines, and we don’t even know which populations the lines came from,” she says. She argues that assaying patterns of X-inactivation across populations and within individuals is one of the challenges the field needs to address before it can fully understand disease phenotype variation.
Interestingly, understanding the evolution of sex chromosomes may shed some light on patterns of variation in X inactivation. One of the topics Wilson Sayres discussed at the meeting was how gene loss on the Y chromosome influences escape from silencing on the X.
In the evolutionary past, the X and Y chromosomes were homologous, carrying mostly the same set of genes. But over time, the Y has lost many genes, leaving it much smaller than the X. Wilson Sayres set out to test the idea that X inactivation of a gene only evolves when the gene’s homolog on the Y (technically a “gametolog”) is lost. Silencing one copy of the gene in females would balance out the mismatch between the sexes.
If this is the case, genes on the X chromosome were once expressed from both copies in females (i.e. they “escaped” silencing), but only became subject to X inactivation when their Y gametolog lost function. By re-examining the X inactivation data from the nine cell lines derived from different people, Wilson Sayres and colleagues confirmed that X linked genes with a Y gametolog escape silencing. In contrast, genes with no gametolog on the Y were, on average, silenced in most cell lines.
Crucially, genes with non-functional gametologs—which are earlier along in the process of being lost from the Y—escaped silencing at a greater rate than those that had completely lost the Y gametolog. That suggests an evolutionary lag between loss of the gene on the Y, and the onset of silencing of the gametolog on the X. This model predicts that the genes that still “escape” silencing even though their Y gametolog is no longer function, will eventually be silenced. “This process is still happening in us,” says Wilson Sayres. “There’s ongoing evolution.”
One implication of this continuing adjustment is that different species are at different stages in the evolution of X inactivation. The process is more tightly regulated in mice, perhaps because the mouse Y chromosome has lost comparatively more of its genes than the human Y. A much smaller proportion of genes escape silencing, and there may be lower variation between individuals.
“It might actually be less messy in mice than in humans,” says Wilson Sayres. This has implications for mouse models of human disease, particularly in cases where the phenotype is modified by genes on the X chromosome.
Solve for X
There is much work to do, Keinan and Wilson Sayres agree. Keinan says researchers need to re-analyze the 2000 or so available GWAS, not to mention the newer sequence-based datasets, most of which still do not consider X. He wonders what proportion of the infamous “missing heritability” of complex human traits might be explained by X-linked variants, but more importantly, hopes we can pin down specific variants and pathways that contribute to the sexual dimorphism of human disease.
Wilson Sayres argues that we need to survey human variation in X-inactivation not only across populations, but within individuals, to get a handle on the patterns of tissue mosaicism in X silencing. She would also like to see more diversity incorporated into association studies. One example would be to include individuals with sex chromosome aneuploidies, instead of excluding them from analysis.
“In Turner Syndrome — that’s where you have only a single X — there’s huge variation in phenotype. Some people don’t even know they have it until they do a cheek swab,” she says. “A huge challenge in studying sex-chromosome aneuploidies is that we don’t understand X variation in karyotypically-common people, let alone for those with single or multiple copies.”
But that’s for the long term, she says. “The first step is just to include the X!”
Cristy Gelling is a science writer, lapsed yeast geneticist, and former Communications Director at the GSA.
View all posts by Cristy Gelling »Read more in
-

Early Career Leadership Spotlight: Julio Molina Pineda
We’re taking time to get to know the members of the GSA’s Early Career Scientist Committees. Join us to learn more about our early career scientist advocates. Julio Molina Pineda Policy and Advocacy University of Arkansas Research Interest My research interests focus on using model organisms to genetically dissect complex traits related to human disease. My…
-

Early Career Leadership Spotlight: Peiwei Chen
We’re taking time to get to know the members of the GSA’s Early Career Scientist Committees. Join us to learn more about our early career scientist advocates. Peiwei Chen Accessibility Subcommittee California Institute of Technology Research Interest Far from a harmonious place, the genome is a battleground, where every bit of DNA fights for inheritance and…
-

#Dros23 GSA Poster Award winners
We are pleased to announce the GSA Poster Award winners from the 64th Annual Drosophila Research Conference! Undergraduate and graduate student members of the GSA were eligible for the awards, and a hard-working team of postdocs volunteered their time as judges. Congratulations to all! Undergraduate Students 1st Place: Sofia Karter Lopez, University of Toronto “Rab11 mediates E-cadherin recycling during…
-

Congratulations to the Fall 2022 DeLill Nasser Awardees!
GSA is pleased to announce the recipients of the DeLill Nasser Award for Professional Development in Genetics for Fall 2022! Given twice a year to graduate students and postdoctoral researchers, DeLill Nasser Awards support attendance at meetings and laboratory courses. The award is named in honor of DeLill Nasser, a long-time GSA supporter and National Science Foundation…
-

New editors join GENETICS, G3 editorial boards
Several new editors are joining the GSA Journals. We’re excited to welcome Ricardo Zayas to the GENETICS editorial board under the Molecular Genetics of Development section, and on the G3: Genes|Genomes|Genetics board, we welcome Polly Campbell, Kevin Vogel, Joe Parker, and Ricardo Mallarino. Ricardo Zayas Associate Editor Ricardo Zayas is a Professor of Biology at…
-

Worms and Flies Provide Key Clues to Medical Mystery
This article is part of a series of posts outlining the history and impact of research in experimental organisms. The series is developed in collaboration with the GSA Public Communications and Engagement Committee. By the time Bertrand Might was six months old, it was clear something was amiss. His muscles weren’t developing normally; he was…
-

Congratulations to the 2023 Early Career Leadership Program Cohort!
The Genetics Society of America (GSA) is excited to announce the latest cohort of student, postdoc, and early-career research leaders joining the Early Career Leadership Program. Participants receive training and mentoring while serving on committees charged with understanding the needs, interests, concerns, and challenges of early career scientist members of the GSA. As part of…
-

GSA LOCI: Local Outreach Community Initiatives @ GSA Conferences
Highlights: Local Outreach Community Initiatives (LOCI): The Genetics Society of America is committed to supporting the communities of the host cities of our conferences. This new year, we are excited to reconnect with our GSA community in meaningful ways within and beyond our existing programming. The GSA membership has created a caring and supportive environment…
-

New members of the GSA Board of Directors: 2023–2025
We are pleased to announce the election of five new leaders to the GSA Board of Directors: 2023 Vice President/2024 President Mariana Wolfner Distinguished Professor of Molecular Biology and Genetics and Stephen H. Weiss Presidential Fellow My research has focused on the genes and pathways that mediate sexual development and reproduction, primarily in Drosophila. From…
-

Lance David Miller: Lighting Your Own Fire by Finding the Right Resources
By Daniel J. Gironda In the Paths to Science Policy series, we talk to individuals who have a passion for science policy and are active in advocacy through their various roles and careers. The series aims to inform and guide early career scientists interested in science policy. This series is brought to you by the…
-

Graça Almeida-Porada: The Importance of Communication in a Technologically Advancing World
By Daniel J. Gironda In the Paths to Science Policy series, we talk to individuals who have a passion for science policy and are active in advocacy through their various roles and careers. The series aims to inform and guide early career scientists interested in science policy. This series is brought to you by the…